
Особенности заболевания
Мышечная дистрофия Дюшенна (МДД) является редким генетическим заболеванием и одной из наиболее распространенных форм мышечной дистрофии1. Она характеризуется прогрессирующей мышечной слабостью, которая проявляется в раннем младенческом возрасте, с последующими скелетно-мышечными, дыхательными и сердечно-сосудистыми нарушениями, которые приводят к инвалидности, зависимости от окружающих и ранней смерти2. Причиной МДД являются делеции или мутации в гене DMD, расположенном в хромосоме Xp21 и кодирующем белок дистрофин3. У этого заболевания рецессивный тип наследования, связанный с Х-хромосомой. Однако примерно в 30 % случаев его причиной являются новые мутации3.

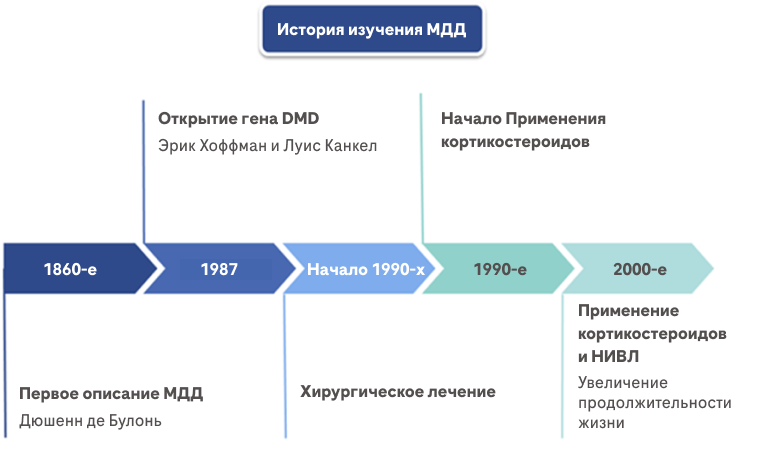
История изучения МДД4
- Французский невропатолог Дюшенн де Булонь был первым, кто подробно описал это заболевание в 60-х годах девятнадцатого века.
- В 1987 г. Эрик Хоффман и Луис Канкел обнаружили, что мутации в гене DMD приводят к нехватке белка дистрофина.
- До начала 1990-х пациенты с МДД получали только самое минимальное лечение. Им проводили хирургические вмешательства для коррекции мышечных контрактур или деформаций позвоночника и стоп.
- Кортикостероиды стали применяться для лечения МДД с начала 1990-х. Они привели к кардинальному улучшению прогноза у пациентов с МДД и с тех пор стали частью стандартного метода лечения этого заболевания.
- С начала 2000-х для пациентов с МДД в большинстве стран стала доступной неинвазивная вентиляция легких. Дополнительное использование респираторной поддержки существенно увеличило продолжительность жизни пациентов с МДД4.
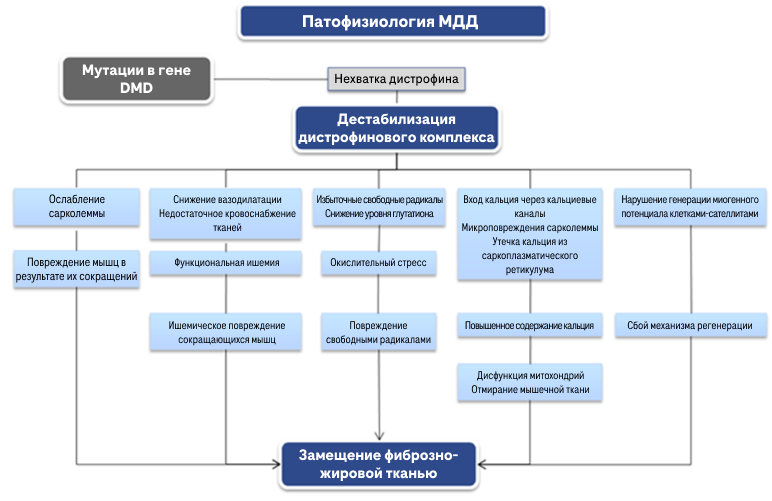
Патофизиология: роль белка дистрофина
Дистрофин экспрессируется клетками всех тканей организма, преимущественно в поперечно-полосатых мышцах (скелетных и сердечной), гладких мышцах, а также в меньшей степени в головном мозге и сетчатке глаза3,5,6. Белок дистрофин локализуется на внутренней поверхности сарколеммы по всей длине мышечного волокна[7]. На поверхности плазматической мембраны мышечных волокон он присутствует в форме дистрофин-гликопротеинового комплекса (ДГК)7. ДГК стабилизирует и укрепляет сарколемму во время мышечного сокращения, защищая таким образом клетки мышц от повреждений, связанных с мышечными сокращениями1,8.
Мутации в генах, кодирующих ДГК, дестабилизируют дистрофиновый комплекс, приводя к слабости и дистрофии мышц1.
Дефицит дистрофина при МДД приводит к следующему9:
- Ослабление сарколеммы. При дестабилизации дистрофинового комплекса возникает ослабление сарколеммы, что делает ее весьма подверженной повреждениям в результате мышечных сокращений.
- Функциональная ишемия. В отсутствие дистрофина снижается вазодилатация, что ведет к недостаточному кровоснабжению тканей и ишемическому повреждению сокращающихся мышц.
- Повреждение свободными радикалами. При МДД свободные радикалы производятся не только воспалительным клеточным инфильтратом и дисфункциональными митохондриями, но также избыточное их количество вырабатывается мышечными клетками. Кроме того, способность мышц противостоять окислительному стрессу уменьшается при снижении уровня глутатиона.
- Повышенное содержание кальция, Уровень кальция в мышцах при МДД выше, чем в мышцах здорового человека. По имеющимся данным, повышенное содержание кальция связано с его усиленным поступлением в мышечное волокно через кальциевые каналы и микроповреждения сарколеммы, а также с его утечкой из саркоплазматического ретикулума, что приводит к дисфункции митохондрий и отмиранию мышечной ткани в результате дегенеративных процессов.
- Сбой механизма регенерации. Дестабилизация дистрофинового комплекса негативно влияет на генерацию миогенного потенциала клетками-сателлитами, нарушая тем самым процессы регенерации.
- Замещение фиброзно-жировой тканью. Все перечисленные выше механизмы запускают формирование дефектных клеточных органелл и аномальных клеточных компонентов, что приводит к дегенерации мышечных клеток. На ранних стадиях заболевания атрофированные мышцы восстанавливаются благодаря механизму регенерации. Однако на более поздних стадиях атрофированные мышечные клетки замещаются жировой и фиброзной тканью из-за нарушения регенераторного потенциала9.
Эпидемиологические данные:
- Распространенность МДД во всем мире составляет 4,8 случая на 100 000 человек5.
- Поскольку ген дистрофина находится в Х-хромосоме, МДД поражает преимущественно мальчиков — на 100 000 лиц мужского пола приходится примерно 7,1 случая заболевания10.
- Распространенность МДД среди новорожденных гораздо выше и составляет 19,8 случая на 100 000 живорожденных детей10. Это означает, что во всем мире МДД поражает 1 из 5000 новорожденных мужского пола6.
- У женщин МДД встречается крайне редко (< 1 случая на миллион), и они чаще всего являются бессимптомными здоровыми носителями9,10.
- Более высокая распространенность МДД среди новорожденных объясняется главным образом тем фактом, что дети с МДД могут не дожить до зрелого возраста, особенно в развивающихся странах10.
- Наибольшая часть мутаций в гене DMD являются делециями (65 %), затем идут дупликации (6–10 %), мелкие мутации (10 %) и другие незначительные генные перестройки7.
Прогноз
Развитие методов симптоматической и поддерживающей терапии, таких как применение глюкокортикоидов, неинвазивная вентиляция легких, кардиопротективная терапия и т. п., привело к увеличению продолжительности жизни пациентов с МДД на 30–40 лет. Тем не менее МДД продолжает оставаться жизнеугрожающим заболеванием, поскольку радикальные способы его лечения пока не найдены8.
-
Узнать больше об МДД:
Список источников:
- Gao Q, McNally EM. The Dystrophin Complex: structure, function and implications for therapy. Comprehensive Physiology. 2015 Jul 7;5(3):1223.
- Osorio AN, Cantillo JM, Salas AC, Garrido MM, Padilla JV. Consensus on the diagnosis, treatment and follow-up of patients with Duchenne muscular dystrophy. Neurología (English Edition). 2019 Sep 1;34(7):469-81.
- Venugopal V, Pavlakis S. Duchenne muscular dystrophy. InStatPearls [Интернет], 11 июля 2022 г. StatPearls Publishing.
- https://www.worldduchenneday.org/history-of-duchenne/. (по состоянию на 16 июня 2023 г.)
- Salari N, Fatahi B, Valipour E, Kazeminia M, Fatahian R, Kiaei A, Shohaimi S, Mohammadi M. Global prevalence of Duchenne and Becker muscular dystrophy: A systematic review and meta-analysis. Journal of orthopaedic surgery and research. 2022 Dec;17(1):1-2.
- Wilton-Clark, H., & Yokota, T. Recent Trends in Antisense Therapies for Duchenne Muscular Dystrophy. Pharmaceutics. 2023;15(3):778.
- Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules. 2015 Oct 7;20(10):18168-84.
- Chey YC, Arudkumar J, Aartsma‐Rus A, Adikusuma F, Thomas PQ. CRISPR applications for Duchenne muscular dystrophy: from animal models to potential therapies. WIREs Mechanisms of Disease. 2023 Jan;15(1):e1580.
- Duan D, Goemans N, Takeda SI, Mercuri E, Aartsma-Rus A. Duchenne muscular dystrophy. Nature Reviews Disease Primers. 2021 Feb 18;7(1):13.
- Crisafulli S, Sultana J, Fontana A, Salvo F, Messina S, Trifirò G. Global epidemiology of Duchenne muscular dystrophy: an updated systematic review and meta-analysis. Orphanet journal of rare diseases. 2020 Dec;15:1-20.