Вы являетесь медицинским работником? Для полного доступа к медицинской информации войдите или зарегистрируйтесь.
О спинальной мышечной атрофии
Спинальная мышечная атрофия (СМА) – редкое, прогрессирующее, инвалидизирующее нервно-мышечное заболевание, которое вызывает атрофию мышц и связанные с ней осложнения
МКБ: G12. Спинальная мышечная атрофия и родственные синдромы
Спинальная мышечная атрофия (СМА) – редкое, прогрессирующее, инвалидизирующее нервно-мышечное заболевание, которое вызывает атрофию мышц и связанные с ней осложнения.
Признаками СМА являются:
- Гибель мотонейронов — нервных клеток в спинном мозге, которые контролируют движение мышц;
- Прогрессирующая денервация мышц;
- Скелетно-мышечная атрофия;
- Общая слабость;
- Потеря двигательной функции.
В зависимости от типа СМА, физическая сила человека и его способность ходить, есть или дышать могут быть значительно уменьшены или полностью утрачены. Мышечная атрофия приводит к осложнениям, связанным с заболеванием, которые могут повлиять на выживаемость1,2.
Мышечная атрофия приводит к осложнениям, связанным с заболеванием, которые могут повлиять на выживаемость
Это заболевание является ведущей генетической причиной смертности младенцев и детей раннего возраста и одно из самых распространенных редких заболеваний, поражающее, по разным данным, одного из 6-11 тыс. детей3-5. В России ежегодно рождаются 300 детей со спинальной мышечной атрофией, а всего в нашей стране может насчитываться от 2500 до 4900 человек со СМА6-7.
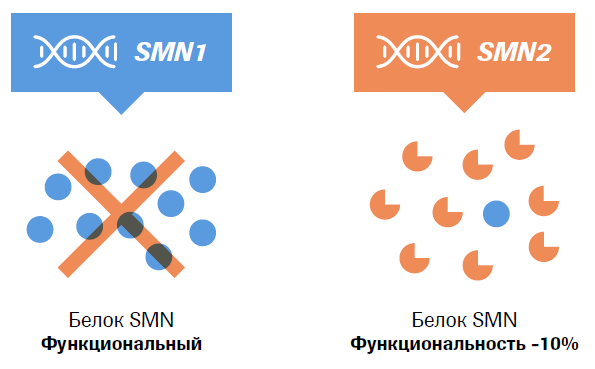
СМА – генетическое заболевание, причиной которого являются мутации или делеции гена SMN1 (survival motor neuron gene), приводящие к недостаточным уровням функционального белка выживаемости мотонейронов (SMN)3.
У гена SMN1 существует псевдоген SMN2, чья кодирующая последовательность отличается от последовательности SMN1 всего одним нуклеотидом (840С>T). Ген SMN2 может частично компенсировать неисправный ген SMN1, путем синтеза небольшого количества (около 10%) функционального белка, необходимого для работы двигательных нейронов.

«Золотым стандартом» молекулярно-генетического исследования при СМА 5q является количественный анализ – анализ числа копий генов SMN1 и SMN2.
- Данное исследование проводится методами ПЦР в реальном времени или количественного MLPA-анализа.
- Диагноз подтверждается при обнаружении делеции экзонов 7 или 7-8 в гомозиготном состоянии (т.е. в обеих копиях гена).
Клиническая классификация заболевания основывается на возрасте дебюта, тяжести течения заболевания и продолжительности жизни.
Дополнительная информация:
МКБ – международная классификация болезней; SMN – (белок) выживаемости мотонейронов; ПЦР – полимеразная цепная реакция; MLPA – разновидность мультиплексной ПЦР, в результате которой амплифицируются несколько различных мишеней только c одной праймерной пары
Ссылки:
- D’Amico A et al. Orphanet J Rare disease 2011;6;71;2.
- Farrar MA et al. Ann Neural 2017;81(3);355-368.
- Lainie Friedman Ross et al. NEOREVIEWS Vol. 20 No. 8 August 01, 2019 pp. e437-e451. Доступ: https://neoreviews.aappublications.org/content/20/8/e437.long.
- Williams L et al. Spinal Muscular Atrophy in the Age of Gene Therapy. AACN Adv Crit Care (2020) 31 (1): 86–91. Доступ: https://doi.org/10.4037/aacnacc2020436.
- Verhaart и соавт., 2017.
- Cure SMA. About SMA. Доступ: www.curesma.org/sma/about-sma.
- Ежегодный бюллетень экспертного совета по редким (орфанным) заболеваниям. Комитет Государственной думы по охране здоровья. Москва, 2020.