Вы являетесь медицинским работником? Для полного доступа к медицинской информации войдите или зарегистрируйтесь.
Классификация СМА (типы и функции)
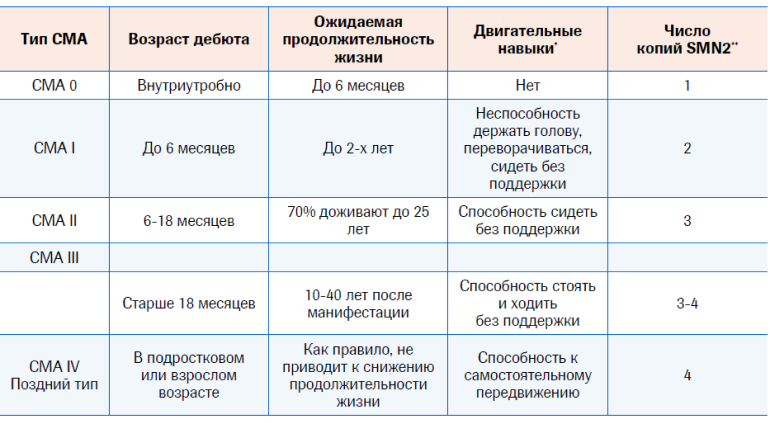
Классическим считается выделение нескольких клинических типов СМА, каждый из которых характеризуется определенным возрастом дебюта, выраженностью клинической картины, максимальными приобретенными двигательными навыками, а также продолжительностью жизни при естественном течении заболевания
Классическим считается выделение нескольких клинических типов СМА, каждый из которых характеризуется определенным возрастом дебюта. Выраженностью клинической картины (тяжесть клинических проявлений заболевания убывает от первого к четвертому типу), что чаще коррелирует с количеством копий гена SMN 2, максимальными приобретенными двигательными навыками, а также продолжительностью жизни при естественном течении заболевания1-3.
Интересно отметить, что число копий гена SMN2 не всегда имеет четкую связь с клиническим фенотипом СМА, т.е. есть существую еще модифицирующие факторы, которые оказывают влияние на дебют и течение заболевания5,6.
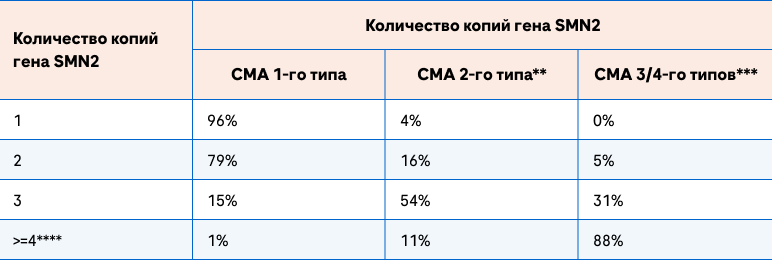
В приеденной таблице прослеживается общая тенденция о том, что чем больше количество копий гена SMN2, тем меньше тяжесть СМА. Вместе с тем, в меньшем проценте случаев возникают исключения. Например, 2 копии гена SMN 2 у пациентов со СМА 3/4-го типов встречаются в 5% случаев, или в 1% случаев у пациентов со СМА 1-го типа обнаружено 4 и более копий гена SMN25,6.

Нулевой тип СМА – дети, которые погибают внутриутробно, либо в первые дни/месяцы жизни в виду выраженной тяжести клинических проявлений.

Тип I заболевания (болезнь Верднига–Гоффманна) является наиболее тяжелым, на него приходится выявление около 60 % больных СМА. Но заболеваемость СМА 1-го типа не отражает распространенности заболевания, которая составляет 12% больных СМА. Это связано с короткой продолжительностью жизни пациентов7. Тип I характеризуется ранним началом, в возрасте до 6 мес, и смертью от дыхательной недостаточности до достижения 2-летнего возраста. Пораженные дети не способны держать голову, переворачиваться, сидеть без поддержки. Проксимальная симметричная мышечная слабость, отсутствие моторного развития и мышечная гипотония – основные клинические признаки СМА типа I. Сохранность диафрагмы в сочетании со слабостью межреберной мускулатуры приводит к формированию куполообразной грудной клетки и парадоксального «брюшного» дыхания, когда ребенок напрягает мышцы живота, а не груди (диафрагма сильнее и используется как основная мышца при дыхании и тянет грудную клетку вниз). Наблюдаются фасцикуляции языка и трудности c глотанием и сосанием, что также способствует снижению защиты дыхательных путей и увеличивает риск развития аспирационной пневмонии – важной причины смерти3,8.

Тип II заболевания, или хроническая СМА (болезнь Дубовица), имеет более позднее начало, в возрасте 6–18 мес., и менее тяжелое течение. Дети сохраняют способность сидеть самостоятельно. Некоторые пациенты могут стоять c поддержкой. Обычно развиваются дислокации тазобедренных суставов, контрактуры, кифосколиоз, которые требуют хирургического или ортопедического вмешательства. Часто наблюдается мелкоразмашистый тремор пальцев вытянутых рук. Слабость глотания может препятствовать набору веса. Как и больные c СМА типа I, из-за нарушения иннервации и слабости межреберной мускулатуры пациенты c СМА типа II испытывают трудности c очищением дыхательных путей от секрета. Дыхательная недостаточность служит частой причиной смерти таких пациентов в подростковом возрасте3,8.

Начало СМА типа III (юношеская форма), или болезни Кугельберга–Веландер, варьируется в возрасте между 18 мес. и первым-вторым десятилетием жизни. Больные со СМА типа III сохраняют способность передвигаться самостоятельно, но часто падают или испытывают трудности при подъеме и спуске по лестнице, беге, наклоне, подъеме из положения сидя. Нижние конечности при данном типе заболевания поражены сильнее, чем верхние. Часто развивается сколиоз3,8.

Также выделяют СМА типа IV. Данное заболевание возникает на 3-й декаде жизни. Характеризуется скрытым началом и медленно прогрессирующим течением. Двигательные нарушения носят мягкий характер, больные не испытывают трудностей c дыханием и питанием, сохраняют способность ходить в зрелом возрасте9.
В настоящее время принято выделять функциональные классы (статусы) пациентов:
- Лежачий/не сидячий;
- Сидячий;
- Ходячий.
Разные клинические типы могут пересекаться в одном функциональном статусе. Например, ребенок (пациент) с третьим клиническим типом, который когда-то приобрел максимальный двигательный навык ходьбу (статус функции – «ходячий»), затем со временем прогрессирования болезни потерял этот навык и перешел в статус «сидячий», а при еще большем прогрессировании – в статус «лежачий». То есть в функциональном статусе «лежачий» теоретически могут пересекаться все три клинических типа заболевания: I тип с самого начала (с момента постановки диагноза), II и III типы при прогрессировании заболевания (при отсутствии должного сопровождения и ухода) с течением времени10.
Дополнительные материалы:
СМА – спинальная мышечная атрофия; SMN – (белок) выживаемости мотонейронов
Ссылки:
- Pearn J.H. Classification of spinal muscular atrophies. Lancet 1980;26;1(8174):919–22.
- Pearn J.H., Walton J.N. A clinical and genetic study of adult-onset spinal muscular atrophy. The autosomal recessive form as a discrete disease entity. Brain 1978;101:591–606.
- Crawford T.O. Spinal Muscular Atrophies. Neuromuscular Disorders of Infancy, Childhood, and Adolescence. A Clinician’s Approach by Jones H.R., De Vivo D.C. and Darras B.T. 2003. Chpt. 8. P. 145–166.
- Crawford T.O., et al. PLoS One. 2012; 7:4
- Calucho M., Bernal S., Alías L. et al. Correlation between SMA type and SMN2 copy number revisited: an analysis of 625 unrelated Spanish patients and a compilation of 2834 reported cases. Neuromuscul Disord. 2018;28:208–15.
- Prior T.W., Swoboda K.J., Scott H.D., Hejmanowski A.Q. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A. 2004;130A:307–10.
- SMA Foundation. http://www.smafoundation.org/wp-content/uploads/2012/03/SMA-Overview.pdf. Доступ 09.11.2023.
- Prior T.W., Russman B.S. Spinal muscular atrophy. Gene Review, 2003.
- Tsao B., Stojic A.S. Spinal muscular atrophy. Emedicine, 2009.
- Сайт пациентской организации Семьи СМА. https://f-sma.ru/all-sma/neurology/nevrologiya/. Доступ 09.11.2023.