Вы являетесь медицинским работником? Для полного доступа к медицинской информации войдите или зарегистрируйтесь.
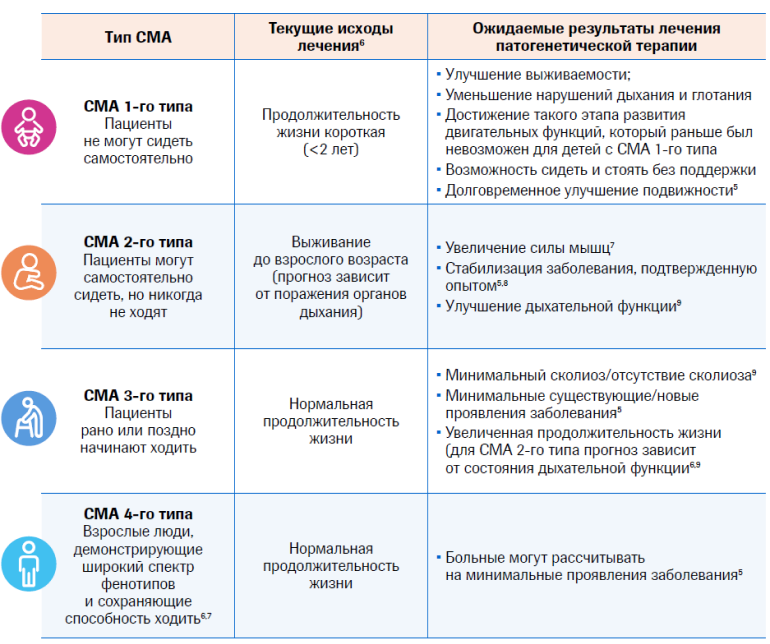
Эволюция фенотипов СМА
Спинальная мышечная атрофия впервые была описана австрийским неврологом Г. Верднигом (Guido Werdnig) в 1891 г.
Долгое время лечение СМА сводилось к поддерживающей терапии или паллиативной помощи. Пациенты с первым, самым тяжелым, типом СМА, как правило, не доживали и до двух лет.
После 2000 года начались активные изучения механизмов СМА на животных, что улучшило знание генетической основы и патофизиологии СМА. Была усовершенствована классификация, в соответствии с которой заболевание СМА было разделено на несколько фенотипов в зависимости от возраста появления симптомов и достигнутой максимальной двигательной функции, которая является наиболее надежным источником для прогнозирования степени тяжести заболевания и продолжительности жизни1.
С 2016 года начала активно внедряться терапия, направленная на коррекцию дефицита белка SMN. Новые методы терапии позволили провести раннее превентивное лечение, которое улучшает выживаемость и сохраняет моторные и респираторные функции пациента2-4.
Новая патогенетическая терапия уже меняет наши ожидания от результатов лечения, а также требует пересмотра и текущая классификация типов СМА, ведь благодаря новым методам лечения теперь можно улучшить клинические симптомы, характерные для каждого типа.
Достижение любого из этапов двигательных функций у разных пациентов будет варьировать и зависеть от различных переменных, включая:5
- Продолжительность болезни
- Уже имеющиеся осложнения
- Функциональный уровень
- Время начала терапии
Согласно Ross LF et al. (2019), в целом эволюцию в подходах ведения пациентов со СМА можно разделить на три периода10:
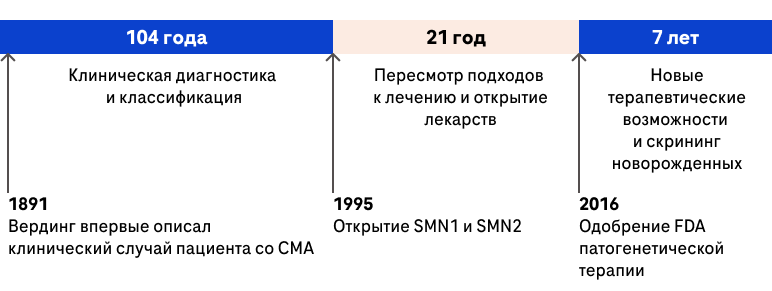
Первый период длился 104 года и начался с момента описания Верднигом клинического случая пациента со СМА в 1891 году. За это время были накоплены знания по течению болезни, разработаны подходы клинической диагностики, выполнен ряд фундаментальных работ, описано применение инструментальных методов исследования, проведен ряд морфологических исследований, сформулирована классификация10.
Второй период длился 21 год и начался в 1995 году с открытия гена SMN1 и SMN2, что послужило катализатором к разработке новых подходов к генетической диагностике и лечению, активно начал применяться мультидисциплинарный подход, открыты новые лекарственные препараты с потенциальным болезнь-модифицирующим действием, проведены клинические исследования потенциальных инновационных методов лечения10.
Третий период начался с 2016 года, когда был одобрен FDA первый препарат патогенетической терапии СМА, за это время разработаны в внедрены в клиническую практику по всему миру три препарата с болезнь-модифицирующий действием (нусинерсен, рисдиплам и онасемногена абепарвовек), появляются новые возможности терапии, начало проведение скрининговых программ среди новорожденных10.
Разработанные современные подходы к ведению пациентов со СМА в третьем периоде повлияют на естественное течение болезни и будут способствовать эволюции фенотипов данного заболевания10.
Дополнительные материалы:
СМА – спинальная мышечная атрофия; SMN – (белок) выживаемости мотонейронов
Ссылки:
- Russman BS, Iannacone ST, Buncher CR, et al. Spinal muscular atrophy: new thoughts on the pathogenesis and classification schema. J Child Neurol 1992;7:347–53.
- Mercuri E et al. Neuromuscul Disord 2018;28:103–115.
- Finkel RS et al. Neuromuscul Disord 2018;28:197–207.
- Kolb SJ, Kissel JT. Neurol Clin 2015;33:831–846.
- Tizzano EF & Finkel RS. Neuromuscul Disord 2017;27:883–889.
- Wang CH et al. J Child Neurol 2007;22:1027.
- Lunn MR et al. Lancet 2008;371:2120–2133.
- Farrar M et al. Ann Neurol 2017;81:355–368.
- Viollet L & Melki J. Handbook of Clin Neuro: Ped Neuro Part III 2013;113:1395–1411. .
- Ross LF, Kwon JM. Spinal Muscular Atrophy: Past, Present, and Future. Neoreviews. 2019 Aug;20(8):e437-e451.